

ISO13485审核重点解析
ISO13485:2016是医疗器械行业专用质量管理体系标准,贯穿器械研发、生产、销售、售后全生命周期,是第三方认证、药监飞检、客户供方审核、海外器械注册核心依据。本文拆解标准基础信息、现场审核核心要点、分级核查内容、高频不符合项与整改误区,搭配表格清晰展示审核重难点,助力器械企业一次性通过内外审核。
汇总注册申报、GMP体系、FDA迎检、客户审计、检测验证和CAPA整改等高频合规主题,帮助企业快速找到可执行的实务参考。
ISO13485:2016是医疗器械行业专用质量管理体系标准,贯穿器械研发、生产、销售、售后全生命周期,是第三方认证、药监飞检、客户供方审核、海外器械注册核心依据。本文拆解标准基础信息、现场审核核心要点、分级核查内容、高频不符合项与整改误区,搭配表格清晰展示审核重难点,助力器械企业一次性通过内外审核。

ISO14971:2019是医疗器械专属风险管理标准,为ISO13485体系强制配套文件,覆盖器械设计、生产、上市后全生命周期风险管控。本文详解标准风控流程、风险判定规则、审计核查要点及常见文件误区,搭配表格梳理风险分级标准,帮助器械企业搭建合规风险管理体系,满足注册与审核要求。

ISO22716是国际通用化妆品良好生产规范标准,适配护肤品、彩妆、洗护等全品类化妆品生产企业,规范车间硬件、人员、物料、生产、检验全流程GMP管控。本文解析标准核心条款、硬性管控要求、客户验厂核查重点、高频整改问题,帮助化妆品工厂顺利通过客户验厂与出口审核。

2026年美国FDA更新药品、医疗器械cGMP规范,重点强化电子数据溯源、实验室质控、OOS偏差管理与供应链审核。本文详解新规改动、核查红线与企业常见违规问题,梳理适配美标的检测管控方案,帮助出口厂商完善检测档案,规避现场核查不合格、产品暂缓进口等合规风险。

原料药、制剂出海企业常会混淆ICH Q7指导原则与各国药品GMP法规,二者适用范围、法定效力、实验室管控要求差异显著。本文从适用对象、核查侧重点、文件管控、第三方检测要求多维度对比,梳理API企业两套标准并行落地方法,规避FDA、EMA现场483缺陷、海外注册受阻等各类合规风险。

药企、原料药厂商搭建FDA认可的cGMP质量体系,需要覆盖美国联邦法规、ICH系列指导原则、电子数据规范、实验室质控、供应链审计多重文件。本文分层梳理建设必备法规清单,拆解各文件核查重点,明确检测、生产、记录全流程落地标准,帮助出口企业一次性搭建完整合规体系。
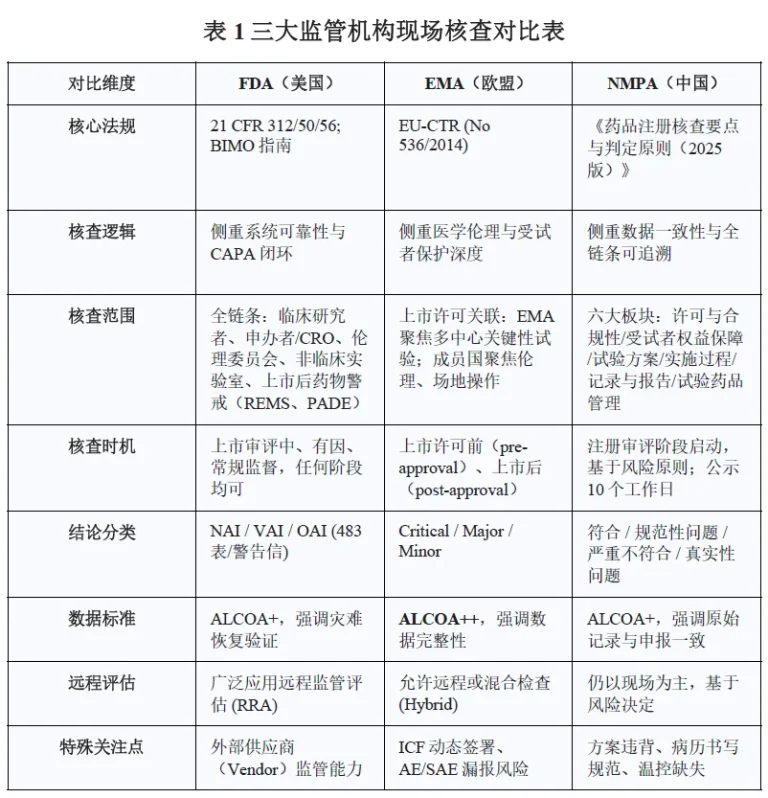
同步布局中美欧市场的医药企业,需同时满足FDA cGMP、EMA EU-GMP、NMPA中国GMP三套规范,三者底层逻辑相近,但数据管控、无菌要求、放行权责、外包检测标准差异明显。本文从法规依据、核查重点、实验室管控、供应链审计多维度对比,为多市场药企搭建兼容型质量体系提供落地参考。

ICH Q7是全球原料药API通用cGMP合规指导原则,适用于化学合成、发酵、植物提取各类原料药生产,是FDA、EMA及国内药监现场审计核心核查文件。本文梳理标准基础信息、适用范围、CMC全流程管控要求、API现场审计核心核查项与判定规则,同时整理原料药企业常见合规误区,助力药企完善质量体系,平稳完成海外注册与官方迎检。

FDA数据完整性是海外药企、原料药、医疗器械企业迎检核心核查项,遵循ALCOA+基本原则,依托21 CFR Part 211法规约束全流程记录。本文拆解数据完整性核心标准、现场审计核查重点、常见不合规缺陷与整改方案,帮助企业完善记录管理体系,规避FDA检查严重缺陷。
提交您的企业类型、产品方向和当前问题,我们将为您匹配相似案例,并提供初步项目建议。
